
Myostatin (GDF-8) x 10mg (1mg x 10 Vials)
$449
10mg Kit
1mg X 10 Vials
All Peptides are shipped non labeled
What is Myostatin (GDF-8)?
Myostatin GDF-8 Peptide is a myokine, a protein produced and released by myocytes that acts on muscle cells to inhibit muscle cell growth. In humans it is encoded by the MSTN gene. Myostatin is a secreted growth differentiation factor that is a member of the TGF beta protein family
Myostatin (GDF-8) Structure
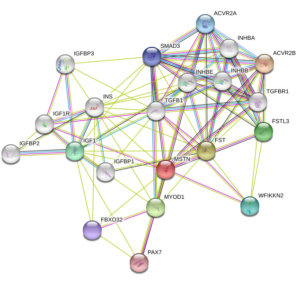
Source: Uniprot
Myostatin (GDF-8) Effects
On Bone Formation
Due to myostatin’s GDF-8 ability to inhibit muscle growth, it can indirectly inhibit bone formation by decreasing the load on the bone. It has a direct signalling effect on bone formation as well as degradation.Knockdown of myostatin GDF-8 has been shown to reduce formation of osteoclasts (multinucleated cells responsible for the breakdown of bone tissue) in mice modeling rheumatoid arthritis. Rheumatoid arthritis is an autoimmune disorder that, among other effects, leads to the degradation of the bone tissue in affected joints. Myostatin GDF-8 has not, however, been shown to be solely sufficient for the formation of mature osteoclasts from macrophages, only an enhancer.
Myostatin GDF-8 expression is increased around the site of a fracture. Suppression of myostatin at the fracture site leads to increased callus and overall bone size, further supporting the inhibitory effect of myostatin GDF-8 on bone formation. One study by Berno Dankbar et al., 2015 found that myostatin deficiency leads to a notable reduction in inflammation around a fracture site. Myostatin affects osteoclastogenesis by binding to receptors on osteoclastic macrophages and causing a signalling cascade. The downstream signalling cascade enhances the expression of RANKL-dependent integrin αvβ3, DC-STAMP, calcitonin receptors, and NFATc1 (which is part of the initial intracellular complex that starts the signaling cascade, along with R-Smad2 and ALK4 or ALK5).
An association between osteoporosis, another disease characterized by the degradation of bony tissue, and sarcopenia, the age-related degeneration of muscle mass and quality have also been found. Whether this link is a result of direct regulation or a secondary effect through muscle mass is not known.
A link in mice between the concentration of myostatin in the prenatal environment and the strength of offspring’s bones, partially counteracting the effects of osteogenesis imperfecta (brittle bone disease) has been found. Osteogenesis imperfecta is due to a mutation that causes the production of abnormal Type I collagen. Mice with defective myostatin were created by replacing sequences coding for the C-terminal region of myostatin with a neomycin cassette, rendering the protein nonfunctional. By crossbreeding mice with the abnormal Type I collagen and those with the knockout myostatin, the offspring had “a 15% increase in torsional ultimate strength, a 29% increase in tensile strength, and a 24% increase in energy to failure” of their femurs as compared to the other mice with osteogenesis imperfecta, showing the positive effects of decreased myostatin on bone strength and formation.
On The Heart
Myostatin GDF-8 is expressed at very low levels in cardiac myocytes. Although its presence has been noted in cardiomyocytes of both fetal and adult mice, its physiological function remains uncertain. However, it has been suggested that fetal cardiac myostatin may play a role in early heart development.
Myostatin GDF-8 is produced as promyostatin, a precursor protein kept inactive by the latent TGF-β binding protein 3 (LTBP3). Pathological cardiac stress promotes N-terminal cleavage by furin convertase to create a biologically active C-terminal fragment. The mature myostatin is then segregated from the latent complex via proteolytic cleavage by BMP-1 and tolloid metalloproteinases. Free myostatin is able to bind its receptor, ActRIIB, and increase SMAD2/3 phosphorylation. The latter produces a heteromeric complex with SMAD4, inducing myostatin translocation into the cardiomyocyte nucleus to modulate transcription factor activity. Manipulating the muscle creatinine kinase promoter can modulate myostatin GDF-8 expression, although it has only been observed in male mice thus far.
Myostatin GDF8 may inhibit cardiomyocyte proliferation and differentiation by manipulating cell cycle progression. This argument is supported by the fact that myostatin GDF8 mRNA is poorly expressed in proliferating fetal cardiomyocytes. In vitro studies indicate that myostatin GDF8 promotes SMAD2 phosphorylation to inhibit cardiomyocyte proliferation. Furthermore, myostatin GDF-8 has been shown to directly prevent cell cycle G1 to S phase transition by decreasing levels of cyclin-dependent kinase complex 2 (CDK2) and by increasing p21 levels.
Growth of cardiomyocytes may also be hindered by myostatin GDF-8 regulated inhibition of protein kinase p38 and the serine-threonine protein kinase Akt, which typically promote cardiomyocyte hypertrophy. However, increased myostatin GDF-8 activity only occurs in response to specific stimuli, such as in pressure stress models, in which cardiac myostatin induces whole-body muscular atrophy.
Physiologically, minimal amounts of cardiac myostatin GDF-8 are secreted from the myocardium into serum, having a limited effect on muscle growth. However, increases in cardiac myostatin can increase its serum concentration, which may cause skeletal muscle atrophy. Pathological states that increase cardiac stress and promote heart failure can induce a rise in both cardiac myostatin GDF-8 mRNA and protein levels within the heart. In ischemic or dilated cardiomyopathy, increased levels of myostatin mRNA have been detected within the left ventricle.
As a member of the TGF-β family, myostatin may play a role in post-infarct recovery. It has been hypothesized that hypertrophy of the heart induces an increase in myostatin as a negative feedback mechanism in an attempt to limit further myocyte growth. This process includes mitogen-activated protein kinases and binding of the MEF2 transcription factor within the promoter region of the myostatin GDF-8 gene. Increases in myostatin levels during chronic heart failure have been shown to cause cardiac cachexia. Systemic inhibition of cardiac myostatin with the JA-16 antibody maintains overall muscle weight in experimental models with pre-existing heart failure.
Myostatin GDF-8 also alters excitation-contraction (EC) coupling within the heart. A reduction in cardiac myostatin GDF-8 induces eccentric hypertrophy of the heart, and increases its sensitivity to beta-adrenergic stimuli by enhancing Ca2+ release from the SR during EC coupling. Also, phospholamban phosphorylation is increased in myostatin-knockout mice, leading to an increase in Ca2+ release into the cytosol during systole. Therefore, minimizing cardiac myostatin GDF-8 may improve cardiac output.
Related Products

CJC-1295 No DAC is a truncated peptide analogue of growth hormone releasing hormone (GHRH). First developed in the 1980s, research studies with modGRF have shown it to improve muscle repair and growth, accelerate wound healing, strengthen bones, increase fat burning, and improve metabolism. It may also have beneficial effects on blood sugar regulation and the immune system.
All Peptides are shipped non labeled
GHRP-6 is a synthetic ghrelin/growth hormone secretagogue agonist. It has positive effects on appetite, heart muscle cells, scar formation, and sexual motivation. Animal studies show this orally active growth hormone secretagogue also improves memory function and may help to thwart the neurological effects of Parkinson’s disease.
All Peptides are shipped non labeled


Out of stock

All Peptides are shipped non labeled
AOD-9604 is a modified version of the hGH fragment 176-191 peptide (contains a di-sulfide bridge) and thus a derivative of human growth hormone (hGH). Originally developed as a lipolytic (fat burning) compound, AOD9604 has shown benefit in studies of heart disease, osteoarthritis/cartilage repair, and metabolic syndrome. AOD9604 stimulates lipolysis (the breakdown or destruction of fat) and inhibits lipogenesis in animal studies.
All Peptides are shipped non labeled



Oxytocin, a natural protein hormone, plays important roles in sexual reproduction, childbirth, bonding between mother and child during breast feeding and wound healing. New research suggests that it may boost cognitive performance, reduce cardiovascular risk, and offset the effects of diabetes.
20mg Kit
2mg x 10 vials
All Peptides are shipped non labeled